
פגיעה בבלוטות אנדוקריניות בעקבות טיפול באימונותרפיה - מדוע שכיח ? ומי בסיכון ?

Introduction
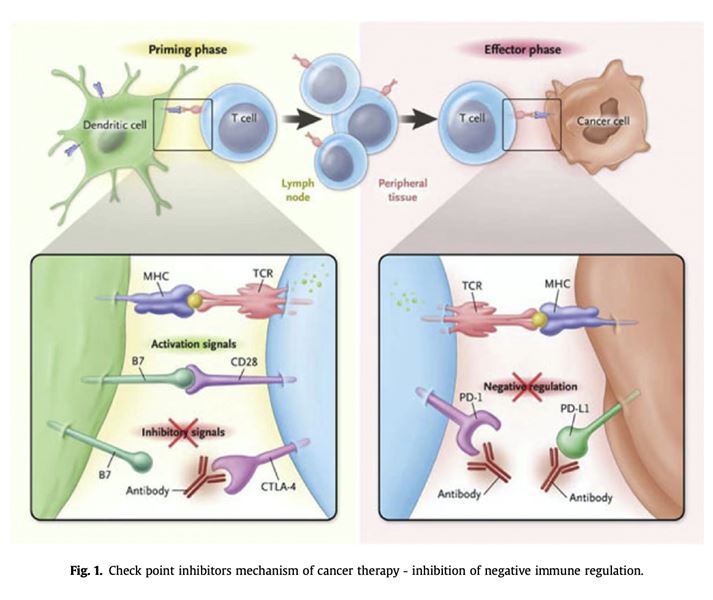
Immunotherapy has transformed the treatment of cancer by restoring the power of the immune system against tumor cells. CTLA-4 blockade supports activation and proliferation of effector T cells and shuts down immunosuppressive regulatory T cells. PD-1 blockade synergistically enhances peripheral T-cell proliferation, function and stimulatory cytokine production [1]. Check point inhibitors (CPIs) have become the standard care in the treatment of a melanoma [2], non-small cell lung carcinoma [3,4], bladder [5,6], renal cell carcinoma [7,8] and more, introducing a dramatic survival benefit (Fig. 1). Disruption of the innate immune system inhibition has introduced a class effect: a large and growing spectrum of autoimmune diseases termed immune-related adverse effects (irAEs) [9e11]. IrAEs induced by CPIs are generally regarded as idiosyncratic, but accumulating evidence indicates that irAEs occurrence depends on a host's propensity for autoimmunity, the type of immunotherapy agent(s) used, and the duration of treatment [10,12]. Some organs are prominent targets to the dys- regulated immune activity, specifically the skin, endocrine and alimentary systems [13e16]. Besides their prominent prevalence, CPI-induced endocrinopathies are characterized by a unique course, being mostly irreversible and unresponsive to glucocorticoid immunomodulation, in contrast to most im- mune related adverse effects (irAE). What makes the endocrine system such a prominent autoimmune target when facing an unleashed immune system? Why are these autoimmune injuries mostly irre- versible and unresponsive to glucocorticoid therapy? Is it possible to identify those prone to develop irAEs in general and endocrinopathies particularly? What are the correlations between irAE and dis- ease response and survival? The present review describes the unique characteristics of the endocrine system and its crosstalk with the immune system in an attempt to shed light on these questions and introduce opportunities for novel research directions. The crosstalk between genetic autoimmune predisposition and immune ignition by check-point inhibition Autoimmune diseases are heavily dependent on genetic propensity depending on genetic loci related to immune regulation, including variations in the HLA-DR system, a class II HLA gene that plays
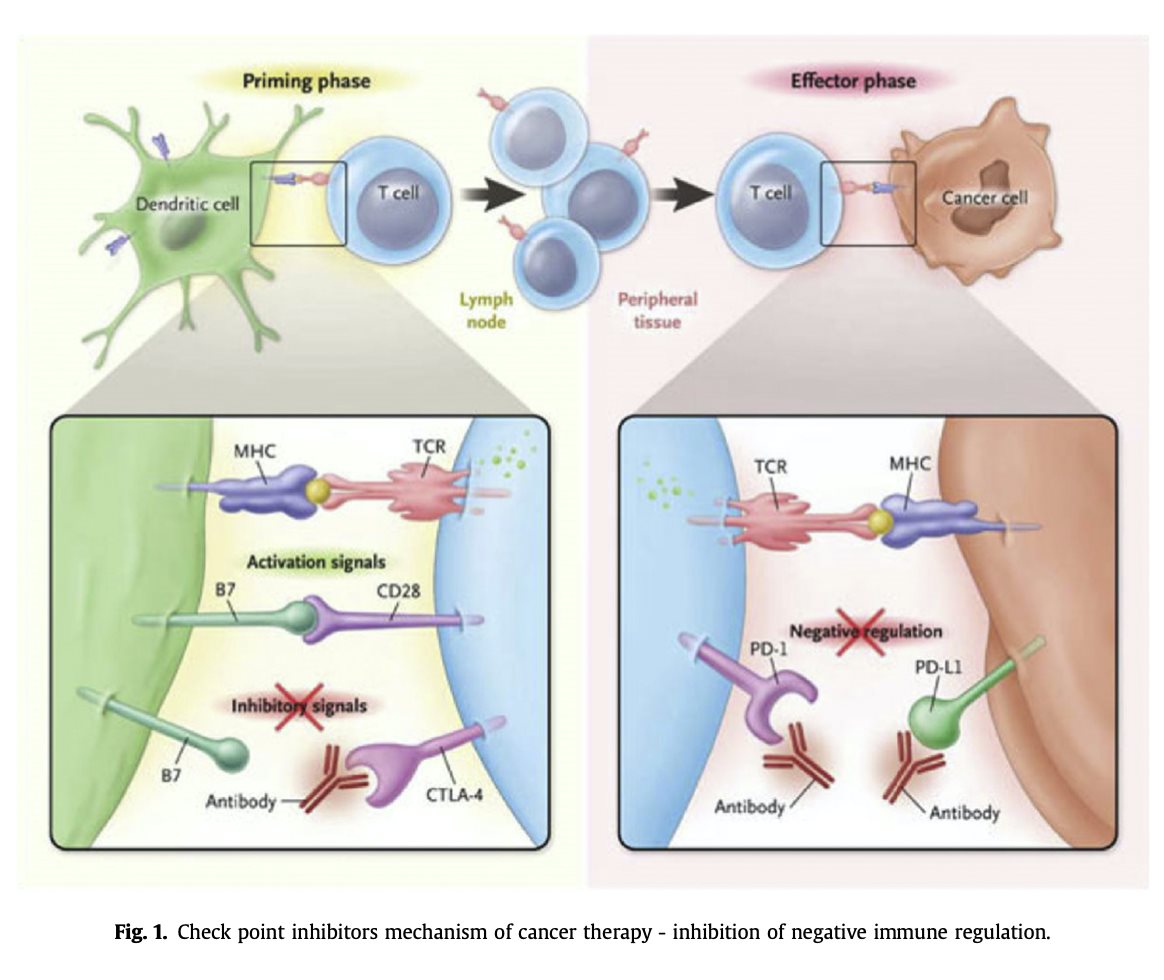
a critical role in antigen presentation. In humans, this genomic locus is known as the human leukocyte antigen (HLA) system, which encodes mostly immune associated proteins whose main role is the presentation of antigens to the immune cells. This system is essential for the proper immune response against pathogens and is strongly implicated in the development of autoimmune diseases. Hundreds of polymorphisms of HLA-DRB1 have been associated with the immune response to infection as well as with different autoimmune disorders [17] as well as with immune response to infection. Recently, the mechanism by which HLA DRB1 haplotypes render an individual to be at a higher risk for autoim- munity was deciphered. These haplotypes, which are associated with an aggressive immune response, possess an evolutionary advantage, i.e.
defending against enteric fever, yet putting them at an increased risk for autoimmunity when an external dysregulation of the immune system occurs [18,19] (Fig. 2).
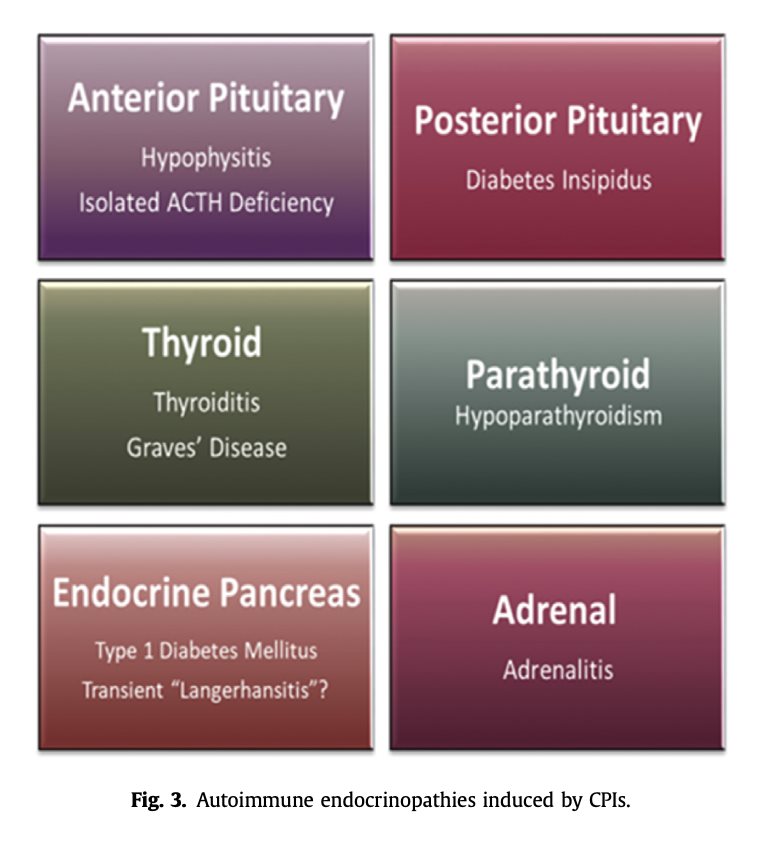
Endocrinopathies induced by check point inhibitors
Thyroiditis is by far the most prevalent and reported irAE, occurring in up to 30% of CPI treated patients [12,20,21] whereas type-1 diabetes mellitus is relatively rare with an estimated prevalence of 0.5e1.9% [9,22e25]. Autoimmune hypophysitis has been predominantly associated with CTLA-4 in- hibition, either alone or in combination with PD-1/PD-L1 inhibition, with a prevalence of about 8% [26,27]. Adrenalitis has been reported scarcely with an estimated prevalence of 0.5% [28] while isolated ACTH deficiency (IAD) causing a secondary adrenal insufficiency with otherwise intact pituitary function, has been recently reported as the predominant CPI induced disease the hypothalamic- pituitary-adrenal (HPA) axis [29e34]. There are rare reports of central diabetes insipidus due to an autoimmune damage to the posterior pituitary [35] and
hypoparathyroidism [36] (Fig. 3).
Why endocrinopathies?
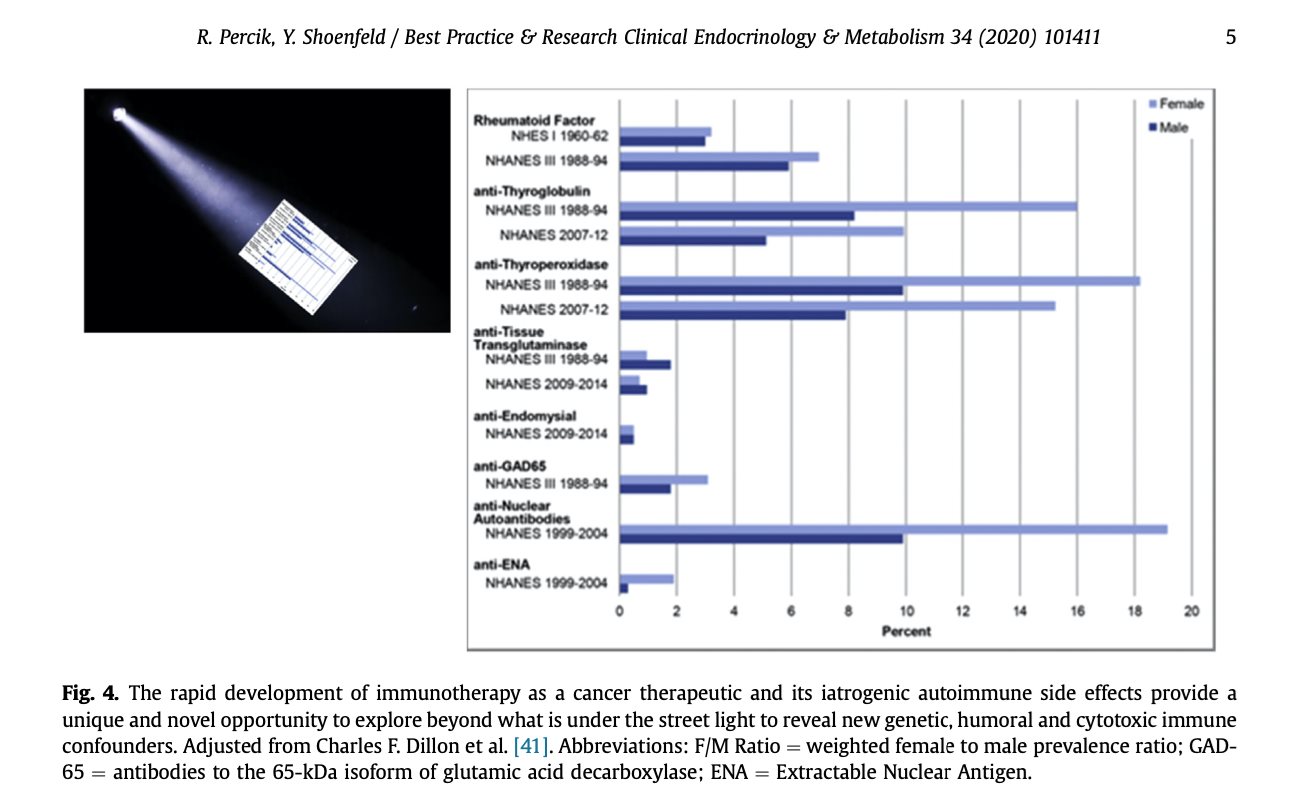
The endocrine system functions in synchronization to regulate hormone production and secretion. As dictated by this physiologic role, the endocrine glands represent a highly communicative network with an intensified signaling load and antigen versatility [37] Self-antigens are conventionally subject to immune tolerance and therefore induce weak or no CD8 T cell responses but bear the potential to magnetize dysregulated T cells provoked by CPIs. Anti-thyroid antibodies are highly prevalent in the
general population (up to 18%), depending on specific antibody, age and gender Even considering the limited laboratory coverage of antibody repertoire in clinical settings and partial correlation between the
presence of autoimmune antibodies and actual autoimmune diseases in general and thyroid dis- ease in particular, the accentuated anti-thyroid antibody repertoire constitutes a fertile soil for cyto- toxic activity in the setting of check point inhibition. The immune ignition pushes humoral thyroid autoimmune propensity to the edge of executive cytotoxicity. The thyroid gland is the most common organ affected by autoimmune disease with an estimated incidence of 350/100 000/year in women and 80/100 000/year in men [38e40]. Autoimmunity to the thyroid, defined by the presence of antibodies to thyroid antigens is reported to be as high as 20% of all women. Autoimmune thyroid disease including chronic, painless and post-partum thyroiditis share a predominately T cell-mediated autoimmunity targeted, partially by the production of thyroid auto- antibodies (Fig. 4). It is assumed that presentation of thyroglobulin and thyroperoxidase peptides by HLA-DR to T cells may be the initial trigger of autoimmune thyroid disease (AITD) both in the general population and among patients treated with CPIs. Genetic studies including genome-wide association studies and whole-genome sequencing and whole-genome linkage screening have led to the identification of several genes whose variants are associated with AITD. These genes function both in immune regu- lation (CTLA-4, HLA genes, PTPN22, FOXP3, CD25, CD40) and as thyroid-specific proteins (thyroglobulin and thyrotropin receptor genes). Specifically, HLADRB1 Arg 74 allele creates a steric structure with possible increased affinity to thyroglobulin as the presented antigen. At last, several CTLA-4 SNPs that have a demonstrated risk for thyroid autoimmunity. Mechanistically, reduced CTLA-4 expression or activity is correlated with decreased suppression of T cell activation and proliferation. The documented 20% prevalence of CPIs induced thyroiditis expresses a complex crosstalk between genetic predisposing determinants and a partially-random interference with the immune system, yet to be explored.
diabetes under check point inhibition express glutamic acid decarboxylase (GAD-65) antibodies, islet cell antibodies (ICA) antibodies, insulin autoantibodies (IAA) or IA-2A antibodies against tyrosine phosphatase [9,22e25]. The correlation between thyroid antibodies and CPI induced thyroiditis is similarly redundant [20,21]. Are there additional autoantibodies to beta cells of the pancreas? What are the antibodies involved in CPI-induced pituitary and adrenal autoimmunity? Are these the known antibodies POMC/ACTH//TPIT/PIT1 and 21-hydroxylase antibodies, respectively [42] or other anti- bodies, yet to be discovered?
The endocrine glands reside on relatively small tissue reservoirs scattered throughout the body and hence, are more vulnerable to an autoimmune attack. Once immunologically damaged, endocrine glands are less likely to regenerate and restore function compared to larger organs. The thyroid gland is a bold example for this principle, as the majority of CPI induced thyroiditis evolve to permanent hy- pothyroidism. In the classic Hashimoto's disease, cytotoxic T cells target the thyroid gland by auto- immune antibodies (anti-thyroperoxidase and anti-thyroglobulin antibodies) and the disease may evolve in various paces and extents: from a partial, intermittent thyroid insufficiency to a complete elimination of thyroid function, depending on the magnitude of cytotoxic recruitment against the thyroid tissue. In contrast, the immune dysregulation induced by CPIs elicits a robust cytotoxic activity leading to rapid and complete tissue consumption in the majority of cases. CPI induced central diabetes insipidus, i.e. autoimmune insult to the posterior pituitary gland and parathyroid injury are irreversible [35,36,43,44], in accordance with proposed reservoirevulnerability correlation. CPI induced diabetes follows the irreversibility rule of endocrinopathies, since the overall size of the pancreas is misleading: the net beta cell mass amounts to 0.8e1.2 g in average [45]. Pituitary and adrenal endocrinopathies, i.e. hypophysitis, isolated adrenocorticotrophic hormone (ACTH) deficiency (IAD) and adrenalitis are challenging to determine whether persistent or potentially transient. Due to the negative inhibition feedback loop of cortisol on the hypothalamus and pituitary and the trophic dependence of the adrenal gland on ACTH, hormone replacement therapy interferes with the possibility for adrenal regeneration [9,46e48]. However, they are mostly considered irreversible. As a whole, CPI induced endocrinopathies evolve more rapidly than the classic autoimmune diseases. A demonstrative difference is reflected in immune related adrenalitis, which evolves within a few months compared to the classic Addison's disease, developing over a 5e20 years' course [49,50]. This difference probably represents the robust and aggressive immune recruitment induced by CPIs.
Better understanding of the complex cross talk between genetic autoimmune determinants and check point inhibition, based on genome-wide association study (GWAS) and whole exome sequencing waits for translation to personalized medicine. A genome based autoimmune predictive matrix will optimize the use of the CPI double edge sword by minimizing the risk for irAEs and maximizing efficacy

*[1] Buchbinder EI, Desai A. CTLA-4 and PD-1 pathways: similarities, differences, and implications of their inhibition. Am J Clin Oncol 2016;39:98e106.
[2] Ott PA, Hodi FS, Robert C. CTLA-4 and PD-1/PD-L1 blockade: new immunotherapeutic modalities with durable clinical benefit in melanoma patients. Clin Cancer Res: Off J Am Assoc Cancer Res 2013;19:5300e9.
[3] Gandhi L, Rodriguez-Abreu D, Gadgeel S, et al. Pembrolizumab plus chemotherapy in metastatic non-small-cell lung cancer. N Engl J Med 2018;378:2078e92.
[4] Garon EB, Rizvi NA, Hui R, et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med 2015;372: 2018e28.
[5] Donin NM, Lenis AT, Holden S, et al. Immunotherapy for the treatment of urothelial carcinoma. J Urol 2017;197:14e22.
[6] Spiess PE, Agarwal N, Bangs R, et al. Bladder cancer, version 5.2017, NCCN clinical practice guidelines in oncology. J Natl
Compr Cancer Netw: JNCCN 2017;15:1240e67.
[7] Motzer RJ, Escudier B, McDermott DF, et al. Nivolumab versus everolimus in advanced renal-cell carcinoma. N Engl J Med
2015;373:1803e13.
[8] MotzerRJ,TannirNM,McDermottDF,etal.NivolumabplusIpilimumabversussunitinibinadvancedrenal-cellcarcinoma. N Engl J Med 2018;378:1277e90.
*[9] Cukier P, Santini FC, Scaranti M, et al. Endocrine side effects of cancer immunotherapy. Endocr Relat Cancer 2017;24:
T331e47.
*[10] Michot JM, Bigenwald C, Champiat S, et al. Immune-related adverse events with immune checkpoint blockade: a
comprehensive review. Eur J Cancer 2016;54:139e48.
[11] Postow MA, Sidlow R, Hellmann MD. Immune-related adverse events associated with immune checkpoint blockade. N
Engl J Med 2018;378:158e68.
[12] Delivanis DA, Gustafson MP, Bornschlegl S, et al. Pembrolizumab-induced thyroiditis: comprehensive clinical review and
insights into underlying involved mechanisms. J Clin Endocrinol Metab 2017;102:2770e80.
[13] BoutrosC,TarhiniA,RoutierE,etal.Safetyprofilesofanti-CTLA-4andanti PD-1antibodiesaloneandincombination.Nat Rev Clin Oncol 2016;13:473e86.
[15] Ntali G, Kassi E, Alevizaki M. Endocrine sequelae of immune checkpoint inhibitors. Hormones 2017;16:341e50.
[16] Albarel F, Castinetti F, Brue T. Management of endocrine disease: immune check point inhibitors-induced hypophysitis.
Eur J Endocrinol 2019;181:R107e18.
*[17] Arango MT, Perricone C, Kivity S, et al. HLA-DRB1 the notorious gene in the mosaic of autoimmunity. Immunol Res 2017; 65:82e98.
[19] Watad A, Rosenberg V, Tiosano S, et al. Silicone breast implants and the risk of autoimmune/rheumatic disorders: a real- world analysis. Int J Epidemiol 2018;47:1846e54.
*[20] OsorioJC,NiA,ChaftJE,etal.Antibody-mediatedthyroiddysfunctionduringT-cellcheckpointblockadeinpatientswith non-small-cell lung cancer. Ann Oncol: Off J Eur Soc Med Oncol 2017;28:583e9.
*[21] Iyer PC, Cabanillas ME, Waguespack SG, et al. Immune-related thyroiditis with immune checkpoint inhibitors. Thyroid: Off J Am Thyroid Assoc 2018;28:1243e51.
[22] Shiba M, Inaba H, Ariyasu H, et al. Fulminant type 1 diabetes mellitus accompanied by positive conversion of anti-insulin antibody after the administration of anti-CTLA-4 antibody following the discontinuation of anti-PD-1 antibody. Intern Med 2018;57:2029e34.
[23] Gaudy C, Clevy C, Monestier S, et al. Anti-PD1 pembrolizumab can induce exceptional fulminant type 1 diabetes. Diabetes Care 2015;38:e182e3.
[24] Clotman K, Janssens K, Specenier P, et al. Programmed cell death-1 inhibitor-induced type 1 diabetes mellitus. J Clin Endocrinol Metab 2018;103:3144e54.
*[25] Lanzolla G, Coppelli A, Cosottini M, et al. Immune checkpoint blockade anti-PD-L1 as a trigger for autoimmune poly- endocrine syndrome. J Endocr Soc 2019;3:496e503.
[26] Joshi MN, Whitelaw BC, Palomar MT, et al. Immune checkpoint inhibitor-related hypophysitis and endocrine dysfunction: clinical review. Clin Endocrinol 2016;85:331e9.
[27] Faje AT, Sullivan R, Lawrence D, et al. Ipilimumab-induced hypophysitis: a detailed longitudinal analysis in a large cohort of patients with metastatic melanoma. J Clin Endocrinol Metab 2014;99:4078e85.
[28] Min L, Ibrahim N. Ipilimumab-induced autoimmune adrenalitis. Lancet Diabetes Endocrinol 2013;1:e15.
[29] Shrotriya S, Rai MP, Alratroot A, et al. Delayed presentation of isolated adrenocorticotropin insufficiency after nivolumab
therapy for advanced non-small-cell lung carcinoma (NSCLC). BMJ Case Rep 2018;2018.
[30] Takebayashi K, Ujiie A, Kubo M, et al. Isolated adrenocorticotropic hormone deficiency and severe hypercalcemia after
destructive thyroiditis in a patient on nivolumab therapy with a malignant melanoma. J Clin Med Res 2018;10:358e62.
[31] Narahira A, Yanagi T, Cho KY, et al. Isolated adrenocorticotropic hormone deficiency associated with nivolumab therapy. J
Dermatol 2017;44:e70.
[32] Escobar-Morreale H, Serrano-Gotarredona J, Varela C. Isolated adrenocorticotropic hormone deficiency due to probable
lymphocytic hypophysitis in a man. J Endocrinol Invest 1994;17:127e31.
[33] TakayaK,SonodaM,FuchigamiA,etal.Isolatedadrenocorticotropichormonedeficiencycausedbynivolumabinapatient
with metastatic lung cancer. Intern Med 2017;56:2463e9.
*[34] PercikR,ShlomaiG,TiroshA,etal.Isolatedautoimmuneadrenocorticotropichormonedeficiency:fromararediseaseto
the dominant cause of adrenal insufficiency related to check point inhibitors. Autoimmun Rev 2020;19:102454.
[35] ZhaoC,TellaSH,DelRiveroJ,etal.Anti-PD-L1treatmentinducedcentraldiabetesinsipidus.JClinEndocrinolMetab2018;
103:365e9.
[36] Win MA, Thein KZ, Qdaisat A, et al. Acute symptomatic hypocalcemia from immune checkpoint therapy-induced hypo-
parathyroidism. Am J Emerg Med 2017;35:1039 e5e7.
[37] KleinL,KyewskiB,AllenPM,etal.PositiveandnegativeselectionoftheTcellrepertoire:whatthymocytessee(anddon't
see). Nat Rev Immunol 2014;14:377e91.
[38] JacobsonDL,GangeSJ,RoseNR,etal.Epidemiologyandestimatedpopulationburdenofselectedautoimmunediseasesin
the United States. Clin Immunol Immunopathol 1997;84:223e43.
[39] Hollowell JG, Staehling NW, Flanders WD, et al. Serum TSH, T(4), and thyroid antibodies in the United States population
(1988 to 1994): National Health and Nutrition Examination Survey (NHANES III). J Clin Endocrinol Metab 2002;87:
489e99.
[40] McGrogan A, Seaman HE, Wright JW, et al. The incidence of autoimmune thyroid disease: a systematic review of the
literature. Clin Endocrinol 2008;69:687e96.
[41] Dillon CF, Weisman MH, Miller FW. Population-based estimates of humoral autoimmunity from the U.S. National Health
and Nutrition Examination Surveys, 1960e2014. PLoS One 2020;15:e0226516.
[42] GubbiS,Hannah-ShmouniF,VerbalisJG,etal.Hypophysitis:anupdateonthenovelforms,diagnosisandmanagementof
disorders of pituitary inflammation. Best Pract Res Clin Endocrinol Metabol 2019:101371.
[43] Piranavan P, Li Y, Brown E, et al. Immune checkpoint inhibitor-induced hypoparathyroidism associated with calcium-
sensing receptor-activating autoantibodies. J Clin Endocrinol Metab 2019;104:550e6.
[44] Trinh B, Sanchez GO, Herzig P, et al. Inflammation-induced hypoparathyroidism triggered by combination immune
checkpoint blockade for melanoma. J Immunother Cancer 2019;7:52.
[45] Saisho Y, Butler AE, Manesso E, et al. beta-cell mass and turnover in humans: effects of obesity and aging. Diabetes Care
2013;36:111e7.
[46] Wang PF, Chen Y, Song SY, et al. Immune-related adverse events associated with anti-PD-1/PD-L1 treatment for malig-
nancies: a meta-analysis. Front Pharmacol 2017;8:730.
[47] FajeA.Immunotherapyandhypophysitis:clinicalpresentation,treatment,andbiologicinsights.Pituitary2016;19:82e92.
*[48] de Filette J, Andreescu CE, Cools F, et al. A systematic review and meta-analysis of endocrine-related adverse events
associated with immune checkpoint inhibitors. Horm Metab Res 2019;51:145e56.
*[49] Betterle C, Scarpa R, Garelli S, et al. Addison's disease: a survey on 633 patients in Padova. Eur J Endocrinol 2013;169:
773e84.
[50] BetterleC,GarelliS,PresottoF,etal.FromappearanceofadrenalautoantibodiestoclinicalsymptomsofAddison'sdisease: natural history. Front Horm Res 2016;46:133e45.